A entrada do algoritmo é uma matriz de distâncias e a saída é uma representação da árvore filogenética obtida pelo algoritmo Neighbor Joining.
Entrada e Saída
Você deve assumir que a entrada é um arquivo com a seguinte cara:
5
Alpha 0.000 1.000 2.000 3.000 3.000
Beta 1.000 0.000 2.000 3.000 3.000
Gamma 2.000 2.000 0.000 3.000 3.000
Delta 3.000 3.000 3.000 0.000 1.000
Epsilon 3.000 3.000 3.000 1.000 0.000
Na primeira linha aparece somente o número n de espécies. Seguem n conjunto de dados. Cada conjunto começa, no início da linha, com um identificador da espécie ocupando, no total, 10 caracteres. Caso o identificar tenha menos do que 10 caracteres, brancos devem ser inseridos. A seguir, seguem n números reais representando as distâncias. Esses números reais podem estar espalhados em várias linhas. Porém, cada um dos n conjuntos começa sempre numa nova linha.
A saída do seu programa deve estar de acordo com uma versão do padrão Newick, descrito abaixo. O padrão recebeu esse nome por ter sido criado durante um jantar no restaurante ``Newick's lobster restaurant'' em 1986.
A descrição do padrão é a seguinte. Vamos supor sempre que a árvore a ser representada tenha pelo menos 3 espécies. Como a árvore produzida pelo Neighbor Joining é sem raiz, todo nó interno está ligado a três outros nós. Escolha qualquer nó interno v da árvore, e imagine esse nó raiz da árvore. Esse nó tem 3 descendentes. A representação da árvore no padrão Newick é a seguinte:
(sub1:dist1,sub2:dist2,sub3:dist3);onde sub1, sub2 e sub3 são as representações das 3 subárvores descendentes de v. Os valores de dist1, dist2 e dist3 são os respectivos comprimentos das ligações. Se alguma das subárvores, digamos sub1, é folha, então sub1 é o identificador da espécie. Caso contrário, sub1 é
(sub11:dist11,sub12:dist12)onde sub11 e sub12 são as representações das 2 subárvores descendentes de sub1, e dist11 e dist12 os respectivos comprimentos das ligações.
A árvore pode ser representada em várias linhas de um arquivo. A quebra de linha pode ocorrer em qualquer lugar, exceto no meio de um identificador ou de um comprimento. A representação deve terminar com um ponto-e-vírgula.
Por exemplo, uma representação da árvore correspondente à matriz de entrada acima é:
(Gamma:1.00000,(Delta:0.50000,Epsilon:0.50000):1.50000,(Alpha:0.50000, Beta:0.50000):0.50000);Um desenho dessa árvore é mostrado na figura abaixo.
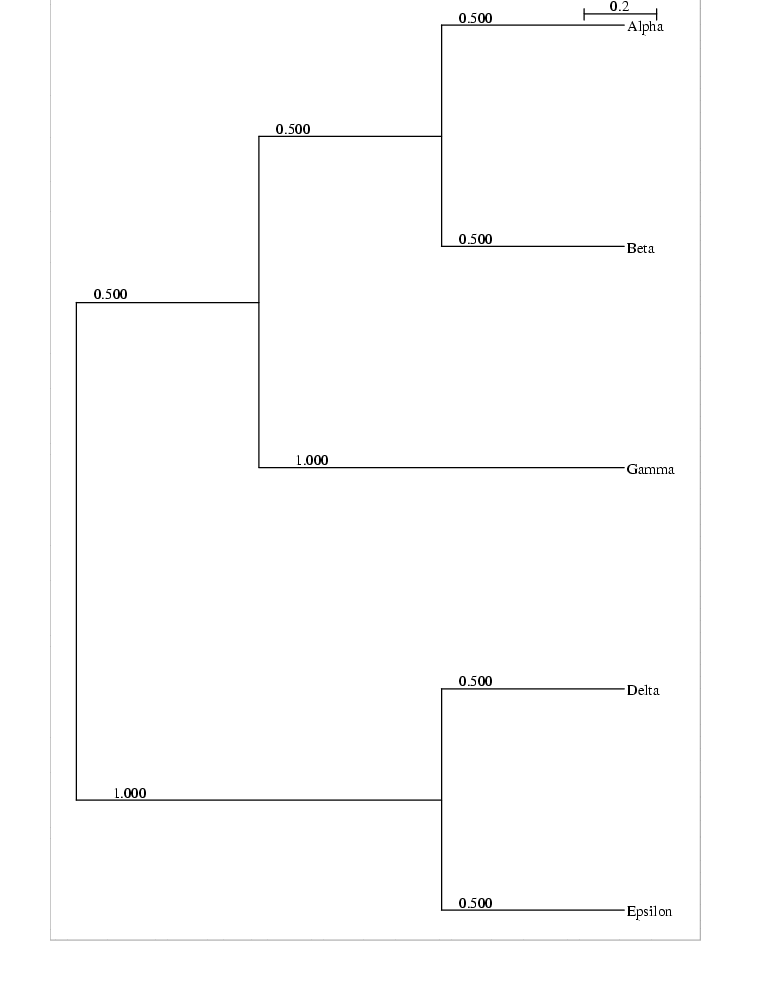
Esse desenho foi gerada pelo programa njplot. Como eu vou testar o seu programa usando o njplot, para facilitar a minha vida, teste a saída de seu programa usando o njplot para se certificar se o formato do arquivo está de acordo com o formato especificado.
Além de outros testes menores, vou testar o seu programa com o arquivo sequencias.dist. (Mas antes, é claro, com entradas menores para entender o que está acontecendo.) Esse arquivo foi gerado a partir de um alinhamento múltiplo das seqüências usando o Clustal. Após a seqüências serem alinhadas, usei o programa dnadist do pacote Phylip para gerar o arquivo sequencias.dist.
No arquivo sequencias.dist alterei a identificação das
seqüências para permitir comparação mais fácil com a árvore obtida no
artigo
Aqui neste arquivo está a representação de uma árvore obtida pelo programa neighbor do pacote Phylip com entrada sequencias.dist.
Programa njplot
Se você estiver na rede do IME numa máquina linux (testei nas máquinas kama e sutra), rode o comandonjplot
Para usar o programa na rede Linux dos alunos, rode o comando
~jose/njplot/njplotNa rede Linux dos alunos você precisa copiar o arquivo ~jose/njplot/njplot.help para o seu diretório de trabalho se quiser que o botão help do njplot funcione.
Outra alternativa é copiar e instalar o programa no seu computador direto do site do njplot.
Outra alternativa é apt-get install njplot para as instalações Debian-linux.
O que entregar
- Seu programa.
- Relatório comentando o trabalho feito, contendo:
- comparação entre a árvore obtida usando como entrada o
arquivo sequencias.dist e a árvore descrita
em
Analysis of the First and Second Internal Transcribed Spacer Sequences of the Ribosomal DNA in Biomphalaria tenagophila Complex (Mollusca: Planorbidae) de T. Vidigal, L. Spatz, J. Kissinger, R. Redondo, E. Pires, A. Simpson e O. Carvalho. - outros comentários sobre a sua implementação.
- comparação entre a árvore obtida usando como entrada o
arquivo sequencias.dist e a árvore descrita
em
 Last modified: Wed Nov 10 18:01:39 EDT 2004
Last modified: Wed Nov 10 18:01:39 EDT 2004
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-